
联系我们
小鼠基因型鉴定详解,必看!
问题一:
Q:鼠达人,实验室新引进新的基因工程小鼠,如何鉴定小鼠的基因型呢?
A:常用的小鼠基因型鉴定方法有:常规PCR鉴定,qPCR鉴定以及测序鉴定,每种鉴定方法都有各自的特点。
01 | 常规PCR鉴定
常规PCR是目前最常用的基因型鉴定方法。通过设计特异性引物扩增目标基因片段(可借助Primer-BLAST等软件设计并校验引物特异性),然后通过琼脂糖凝胶电泳分析PCR产物的条带大小差异来判断小鼠的基因型。
优势:操作简便、成本较低,适用于已知突变位点的基因分型,常见于大多数实验室。
局限性:无法检测未知变异,灵敏度较低。
02 | qPCR鉴定
实时荧光定量PCR(Quantitative Real-Time PCR, qPCR)是一种更为精准的基因型鉴定方法。它通过荧光信号(如SYBR Green或TaqMan探针)实时监测PCR扩增过程,不仅可以判断基因型,还能够通过荧光信号实现基因拷贝数的定量分析。
优势:灵敏度高、特异性强,支持定量分析。
局限性:依赖探针或优化条件,设备成本高,操作复杂度高于常规PCR。
03 | 测序鉴定
在特定研究场景中,测序鉴定(如Sanger测序或NGS)是必不可少的手段,如点突变模型小鼠的基因型验证,或基因编辑产生的微小缺失/插入突变(仅涉及数个碱基)。通过测序可以直接读取目标基因的碱基序列,从而精准判定基因型。
优势:结果更为准确可靠,可发现新突变。
局限性:成本高、数据分析复杂,耗时较长。
问题二:
Q:鼠达人,最常用的是常规PCR鉴定,那具体怎么操作呢?
A:已经准备好了,看这里。
01 | 样本准备
在基因型鉴定中,小鼠基因组DNA的制备是决定实验结果好坏的关键。根据经验,通常在小鼠断奶前后(约3周龄)采集组织用于基因组DNA提取,例如剪取尾尖或耳组织。
不建议在较大龄时剪趾取样,如果必须通过剪趾获取组织,应在小鼠出生后10–15天内完成,以同时起到标记编号作用。
注意:
① 鼠尾尾尖(2~3mm),鼠耳约2~5mm2,脚趾1枚可获得的DNA量足够用于基因型鉴定。
② 剪组织的剪刀每剪完一次需用酒精棉擦拭,避免DNA交叉污染。
③ 避免组织样本带有血液(血液中的溶血产物或抑制剂可能干扰PCR扩增)。
④ 采样后若不立即进行样品消化,需保存于-20℃或更低温度,在消化前避免反复冻融。
02 | DNA提取
1) 将组织(鼠尾)剪碎置于离心管中,加入47 μL裂解液和3μL蛋白酶K(20 mg/mL)。
2) 充分振荡混匀且完全浸没样本后,离心管置于55℃烘箱孵育过夜(确保组织完全消化)。
3) 消化完成的样本,于PCR仪加热至99 ℃处理10 min以灭活蛋白酶K,随后冷却至室温或4℃。
4) 灭活完成的样本加130 μL的ddH2O以稀释DNA,随后混匀,2000 rpm离心1 min将消化剩余的毛发、骨头等固态杂质离心于管底,后续PCR试验取上清作为模板。
5) 提取的DNA产物可短期保存于4 ℃,长期保存可置于-20 ℃或更低温度,避免反复冻融。DNA样品的OD260/280在1.8-2.0之间。
注意:
① 需要充分裂解鼠尾,样本应消化至肉眼看不到颗粒为止。
② DNA模板浓度应控制在50-100ng/μL进行PCR。
03 | PCR扩增
以某双转基因小鼠的基因鉴定方案为例,在进行PCR扩增前,应先确定PCR反应体系和扩增程序。
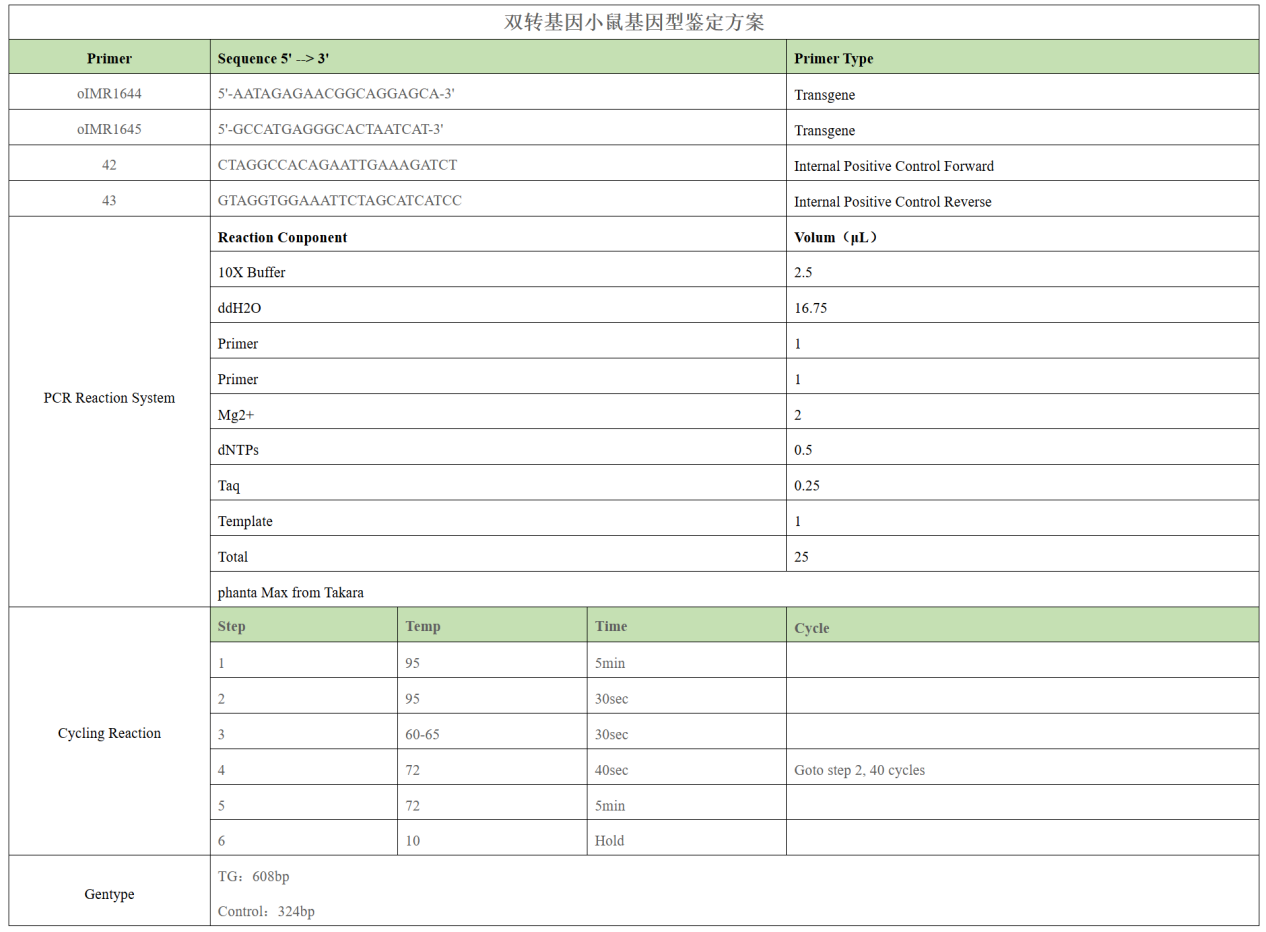
注意:
设置对照Control非常重要,不仅可以帮我们快速鉴别基因型,还能验证PCR反应体系是否正常工作。建议在实验中同时设置:阴性对照(不加模板的反应,确保无污染产生假阳性);阳性对照(含已知基因型的模板,确保引物和反应体系有效)。
04 | 凝胶电泳
1)配置凝胶:根据目标条带配置1%-3%的琼脂糖凝胶,将1g-3g琼脂糖溶于100mL TAE缓冲液,微波炉加热至琼脂糖融化(1min),加入10μL核酸染料(EB替代物),倒入电泳模板中,随后插入梳子,待冷凝后使用。
2)加样:向琼脂糖凝胶中加入适量的DNA,小孔建议加10μL,大孔建议加20μL,并根据目标条带大小选择合适的DNA marker加入凝胶中。
3)跑胶:电泳仪设置100V-120V电压,跑20-30min,以溴酚蓝迁移至胶长2/3处停止。
4)成像:使用凝胶成像系统观察,拍摄照片。
5)结果读取:根据胶图中DNA Marker比对目标条带大小,区分野生型、杂合子及纯合子,出具基因型鉴定结果报告。
注意:
① PCR产物可根据条带大小,选择不同浓度的凝胶进行电泳(200bp:3%;200-1,000bp:1.5%-2%;>1,000bp:1%),后续根据电泳结果进行基因型分析。
② 微波加热时避免暴沸,冷却至60°C再倒胶,减少气泡和孔隙不均。
问题三:
Q:鼠达人,PCR扩增后出现假阳性、假阴性、或者是非特异性条带,怎么解决呢?
A:这些情况在平常操作中,如果没有加以注意,很容易出现,针对此,整理出了以下解决措施供参考。
01 | 假阴性问题
①如果模板中存在微量抑制剂,可适当稀释DNA模板,降低杂质比例;
②重新纯化提取DNA模板(乙醇纯化去除小分子抑制剂、离心柱纯化去除蛋白质/多糖等);
③及时电泳,避免扩增产物长时间存放室温或4°C冰箱导致降解。
02 | 假阳性问题
① 如果是实验过程中出现阳性污染,如试剂耗材污染,建议更换试剂,灭菌耗材;如气溶胶污染,建议UV照射操作环境。
② 如果不是污染造成的,建议重新设计引物进行检测,条带不小于500bp,避免靶序列或引物过短。
03 | 出现非特异性条带
非特异性扩增条带较弱时:
① 降低引物和聚合酶的用量,或者更换酶。
② 提高退火温度,或采用梯度PCR法来优化条件。
③ 降低循环次数,避免引物过多的非特异性结合。
④ 纯化模板或者稀释模版,避免非特异性物质或降解片段的干扰。
非特异性扩增条带较强(非特异性条带的亮度与目的条带几乎相当甚至更亮)时:
建议重新设计PCR引物或尝试使用touchdown PCR。
本期分享就到这里,我们下期再见~
上一页
上一页

实验动物订购电话: 0731-84877202/84877208
广东专线: 400-881-9598
技术服务热线: 0731-84876042
企业QQ: 800073177
邮箱: [email protected]
地址: 湖南省长沙市芙蓉区隆平产业开发区豪丹科技园3栋3楼
关注我们
